
Versatile and Robust Method for Antibody Conjugation to Nanoparticles with High Targeting Efficiency

 ,
,  ,
,  ,
, 
 ,
, 

Abstract
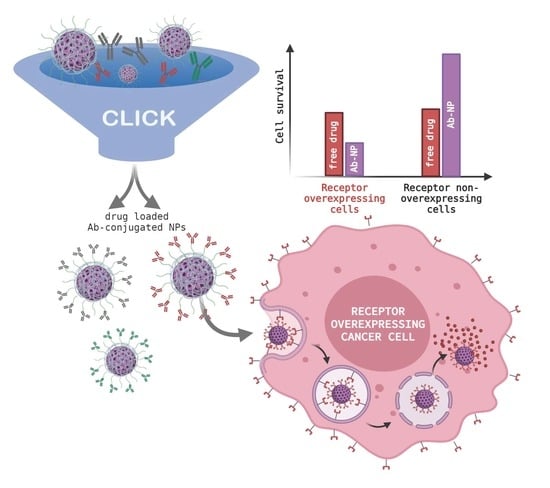
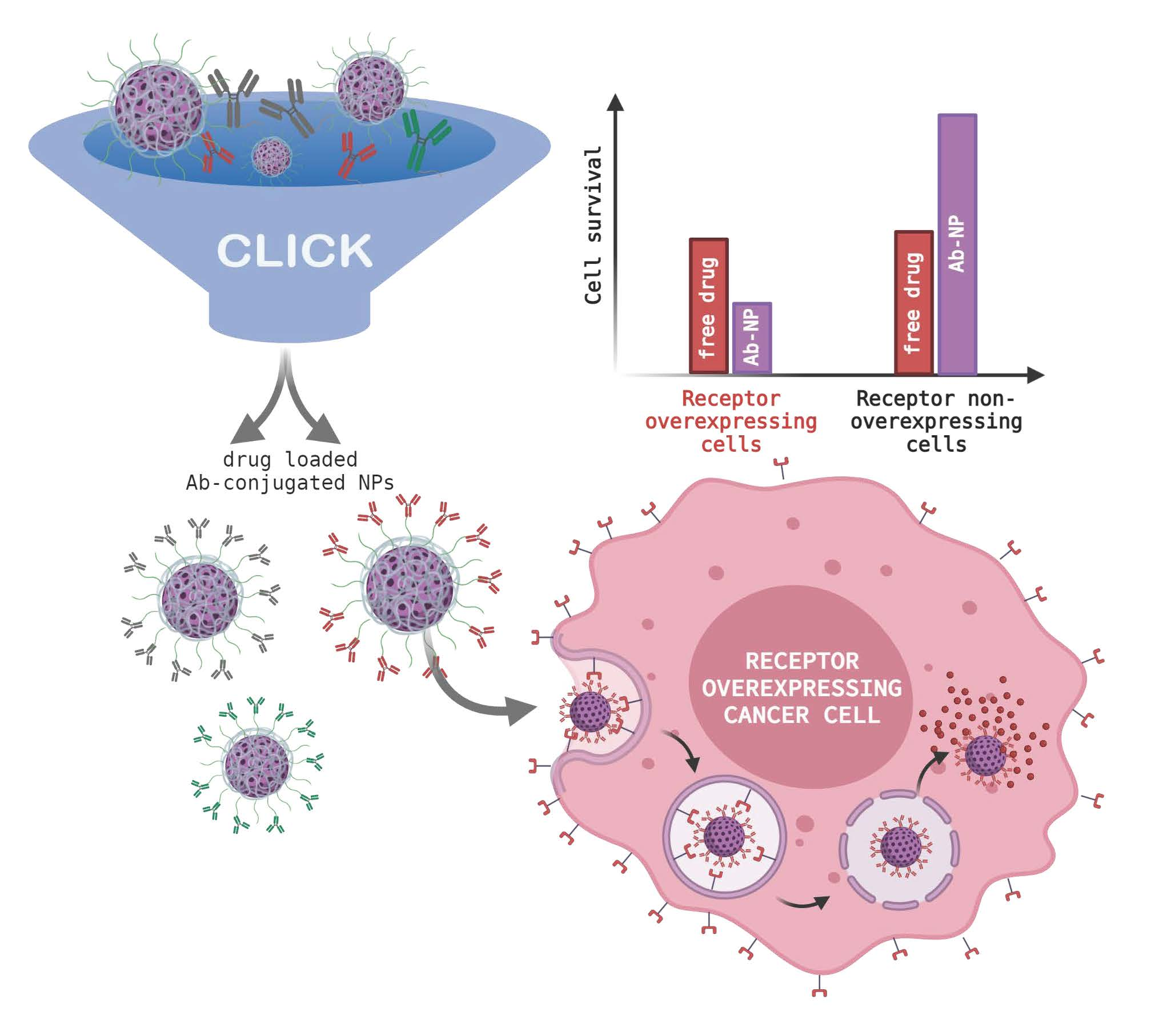
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. CD44, EGFR and IgG Antibody Labelling
2.3. Synthesis MSNPs
2.4. MSNP Dye/Drug Loading
2.5. MSNP Functionalization
2.6. Ab-PEI-MSNPs’ Characterization
2.7. Cell Culture
2.8. Immunofluorescence Labeling
2.9. Ab-PEI-MSNPs’ Targeting Efficiency
2.10. Transfection of Hek293
2.11. Fluorescence Microscopy
2.12. The αCD44-PEI-MSNPs’ Intracellular Localization
2.13. Doxorubicin Release
2.14. Cytotoxicity Studies
2.15. Statistical Analysis
3. Results and Discussion
3.1. The αCD44-Conjugated Nanoparticles
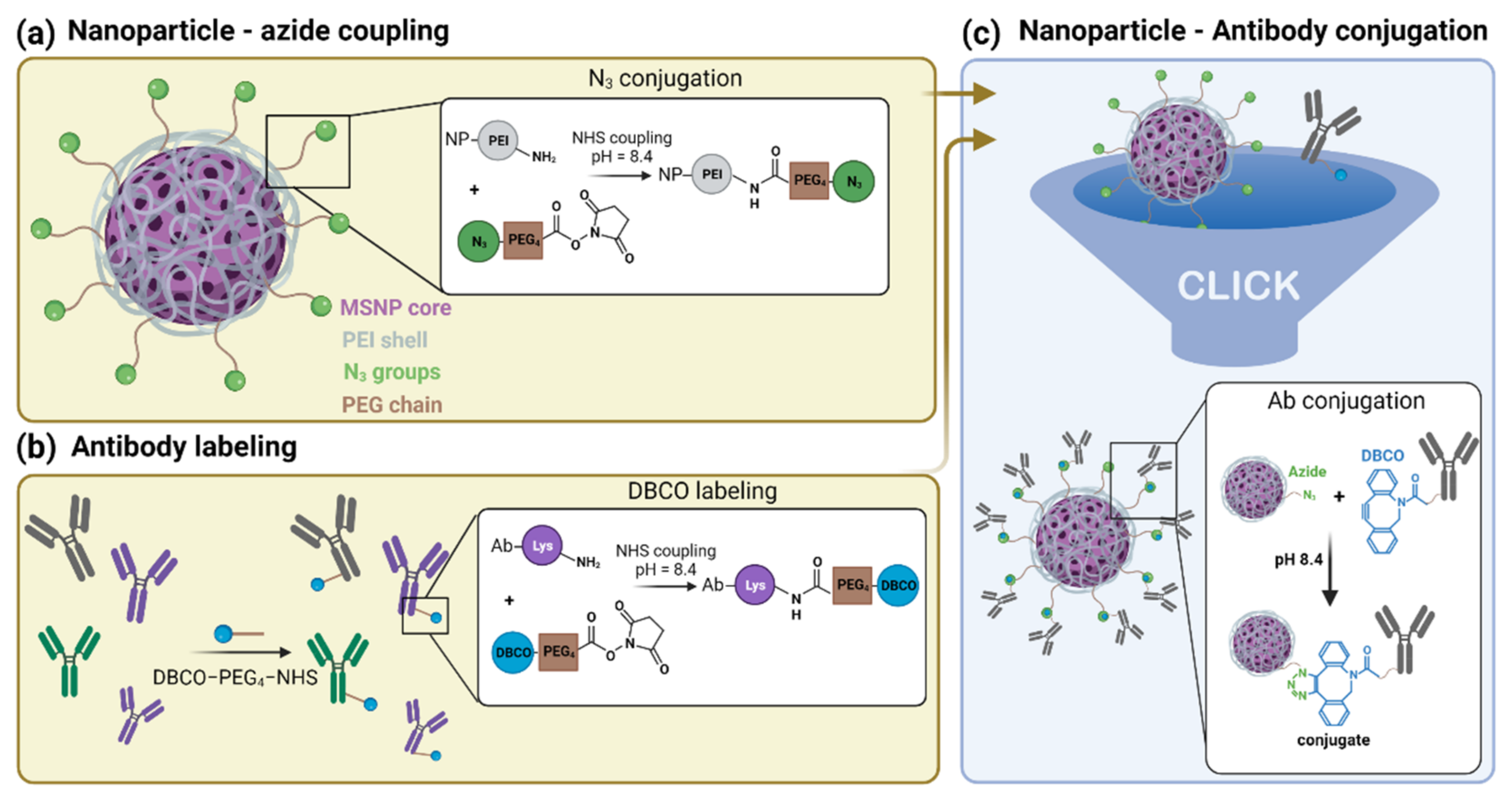
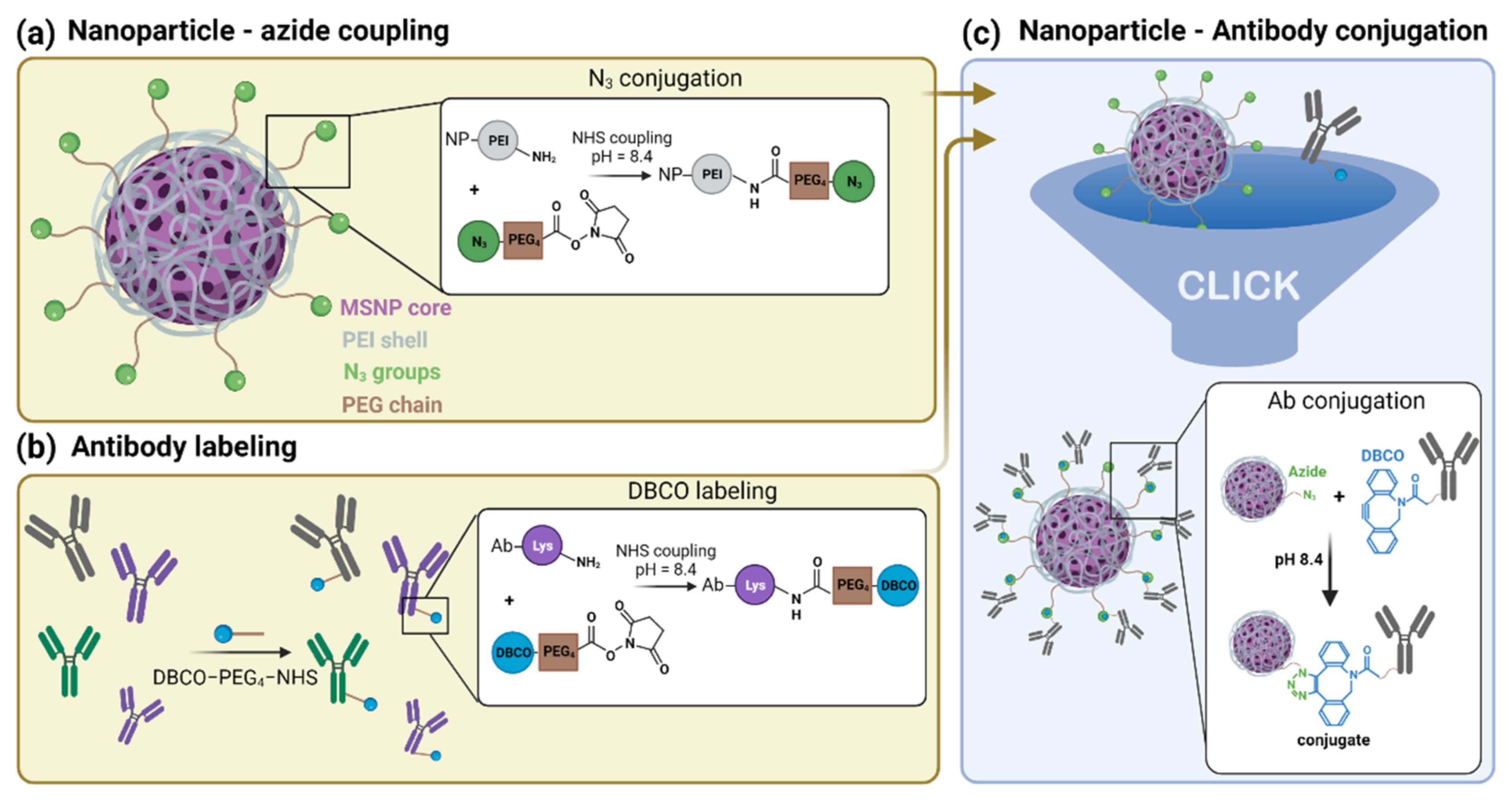
3.1.1. Synthesis of Azide-Functionalized MSNPs
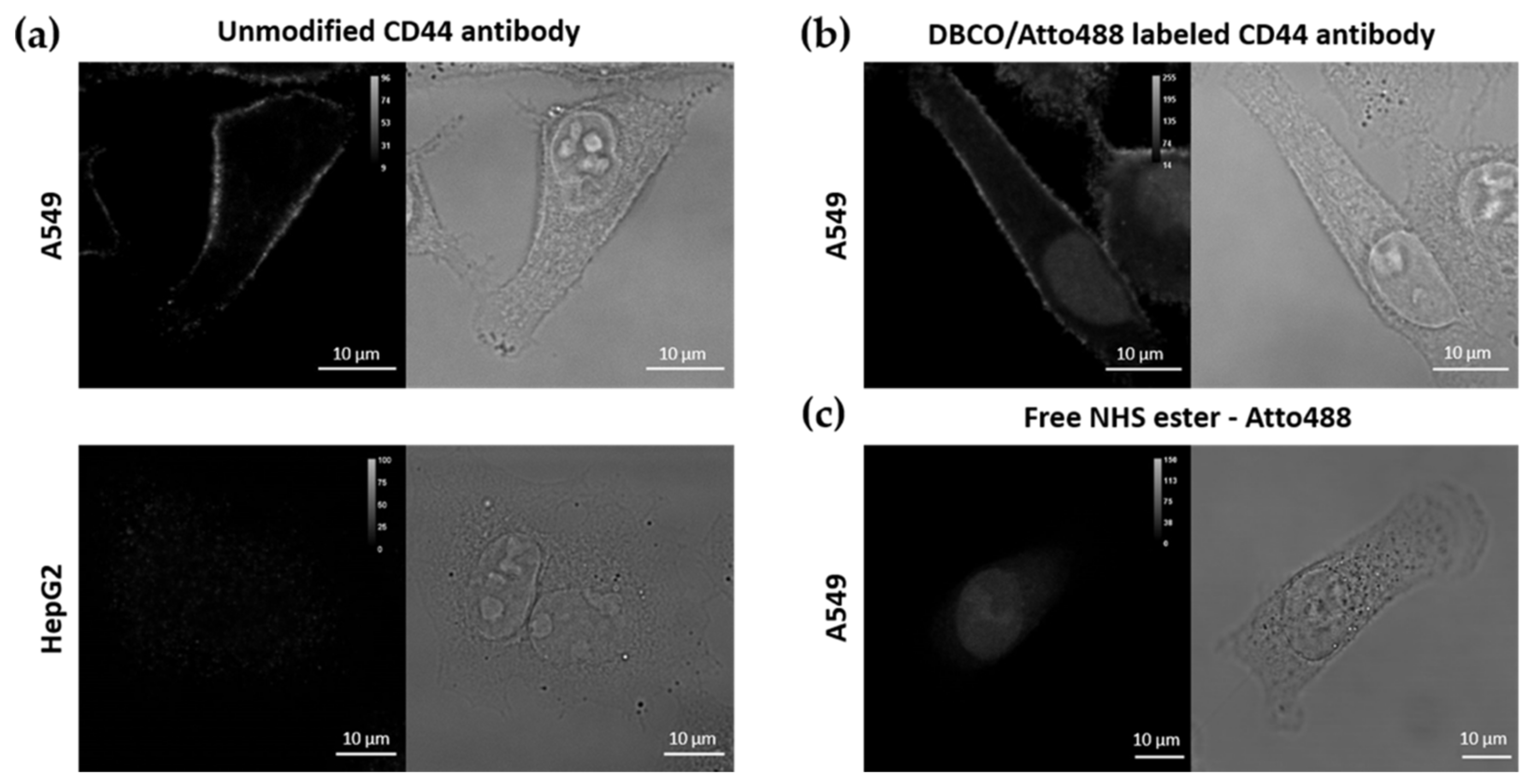
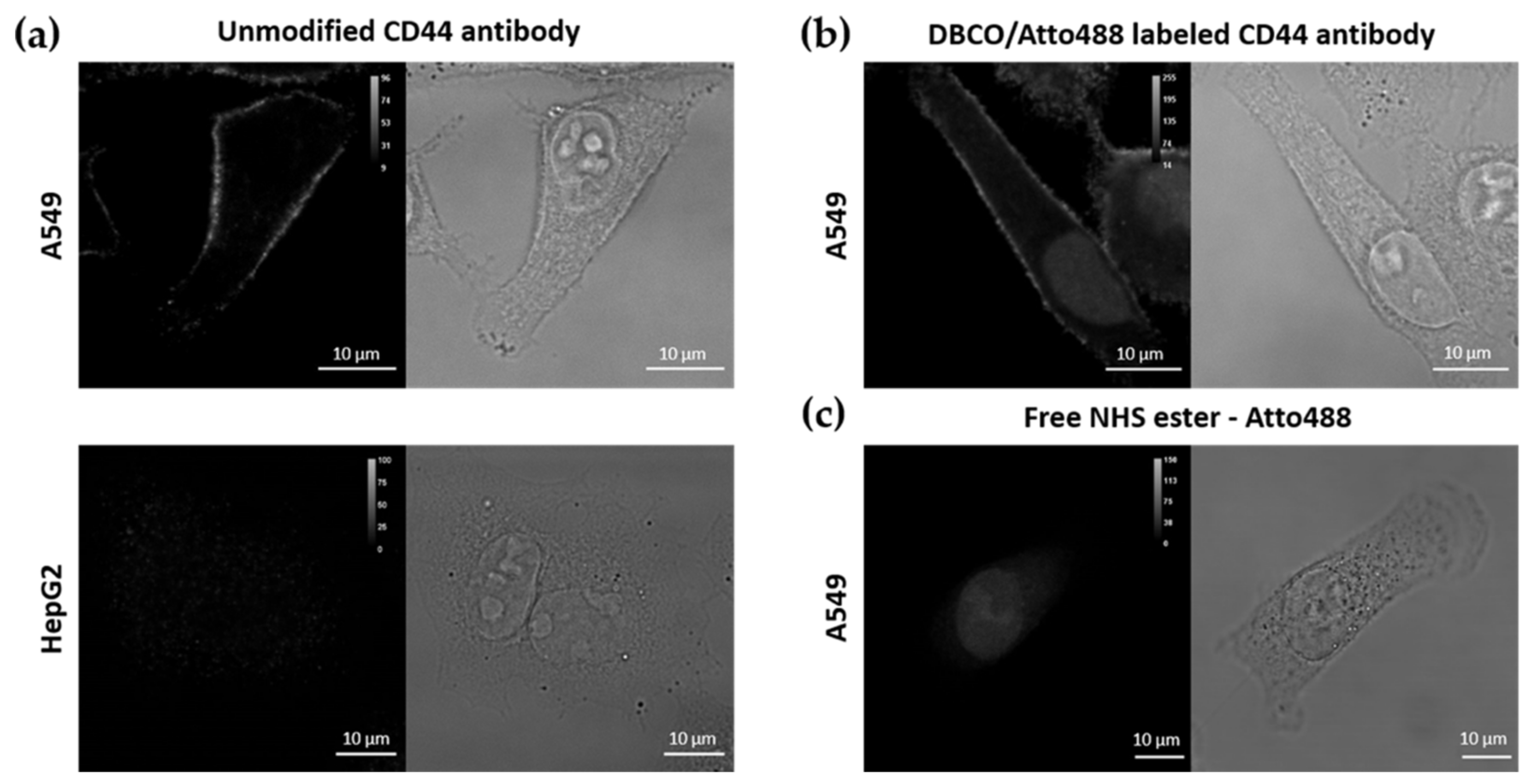
3.1.2. Labelling of CD44 Antibodies
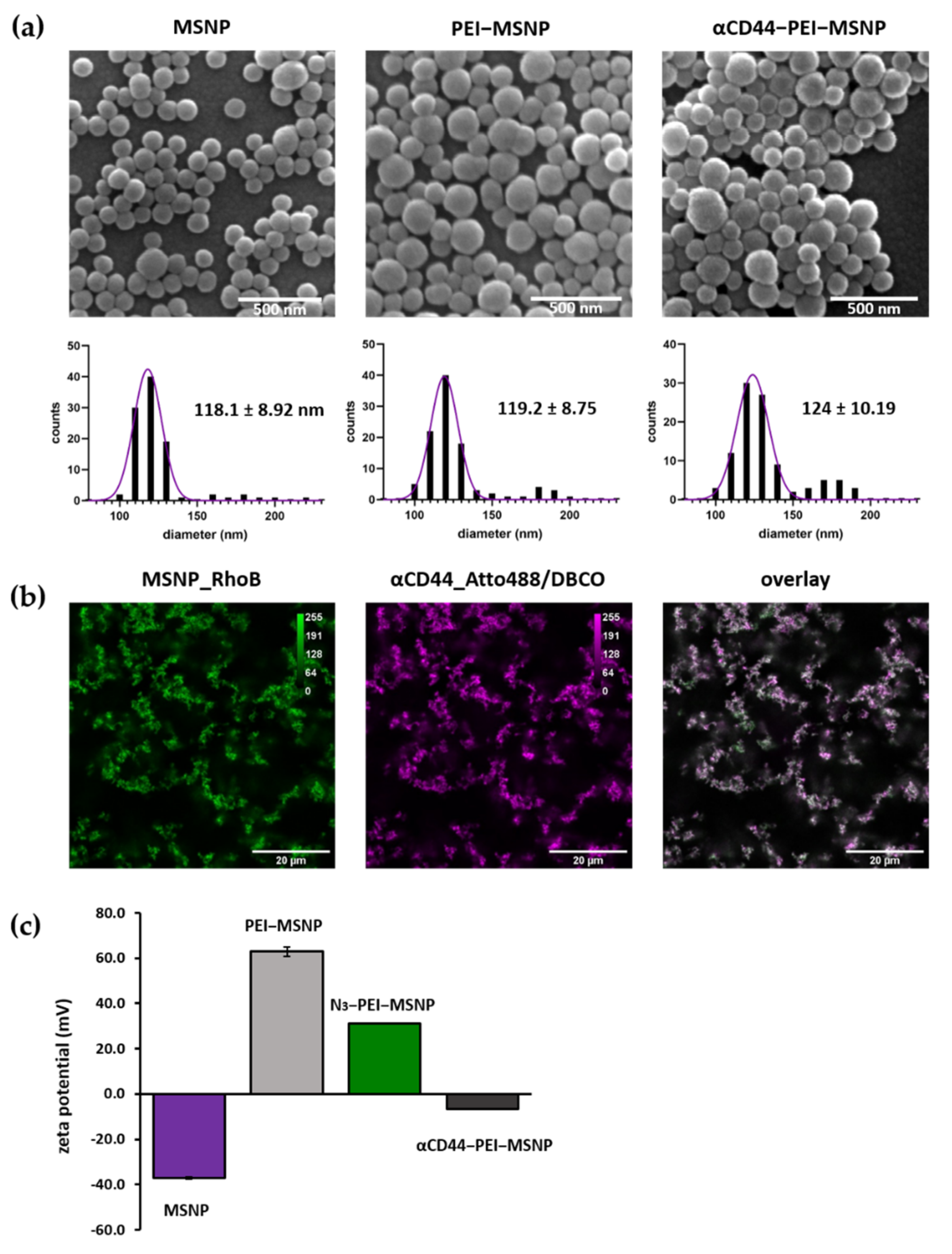
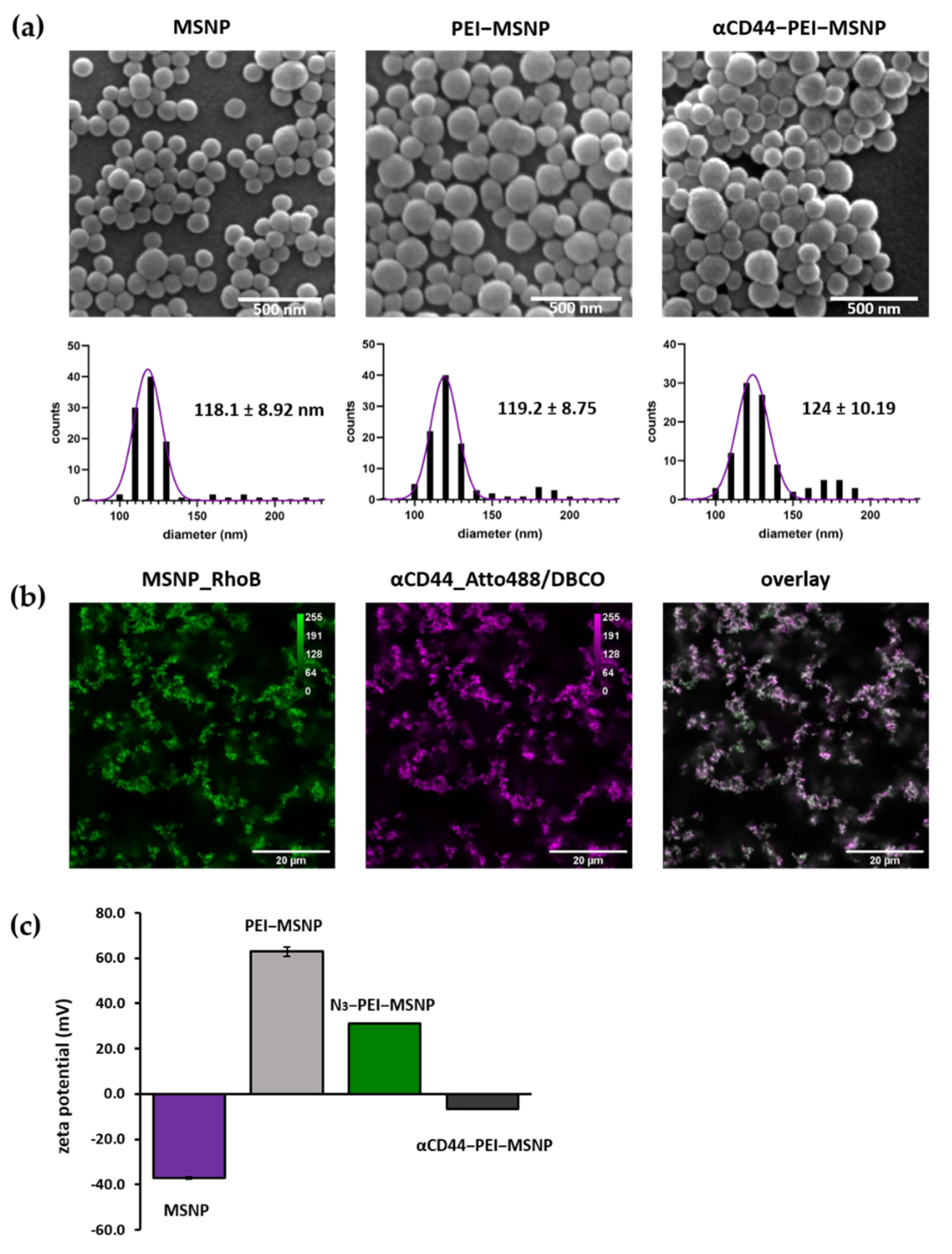
3.1.3. Synthesis and Characterization of αCD44-Conjugated MSNPs
3.2. Selectivity and Efficiency of αCD44-Functionalized Particles
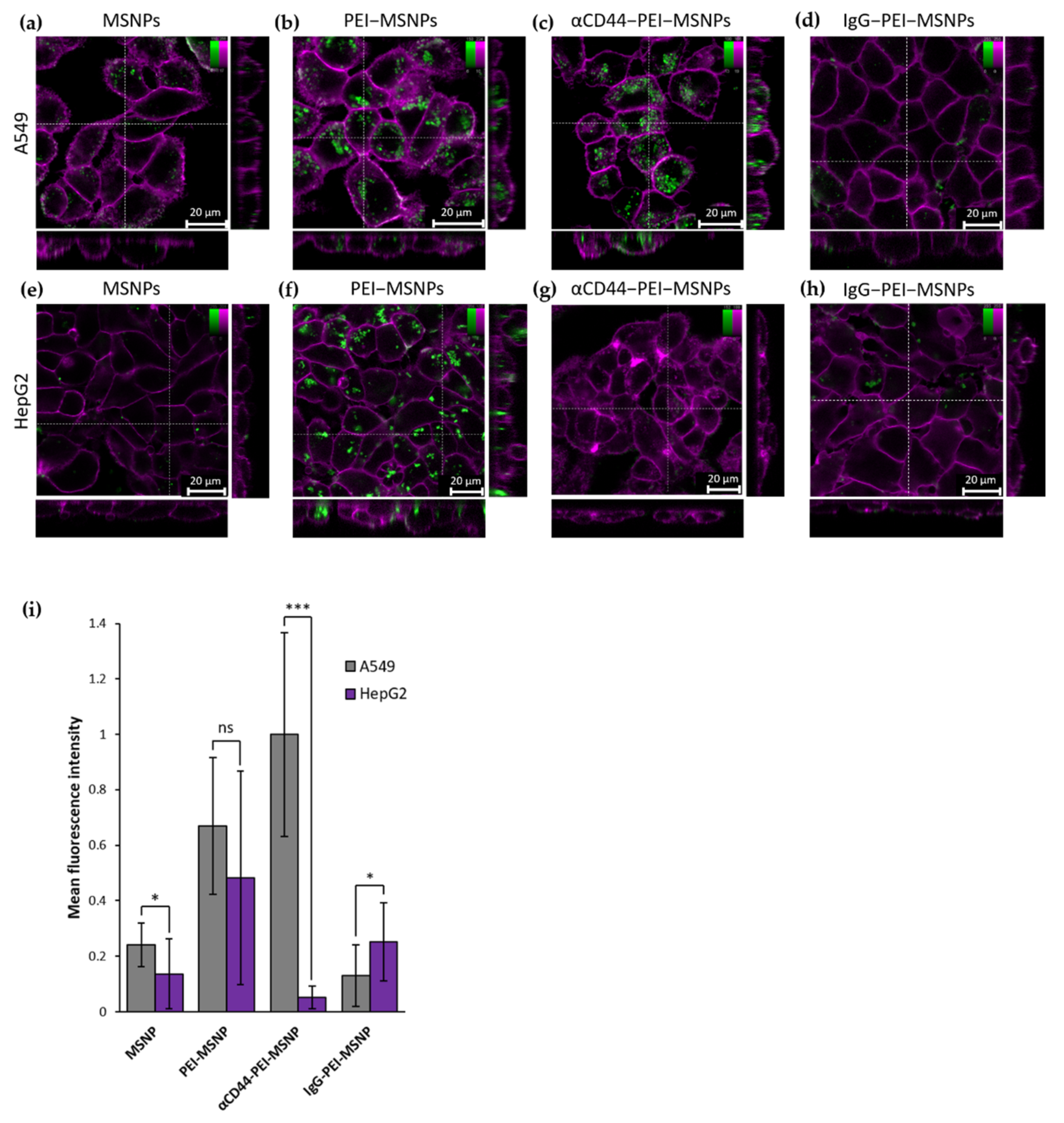
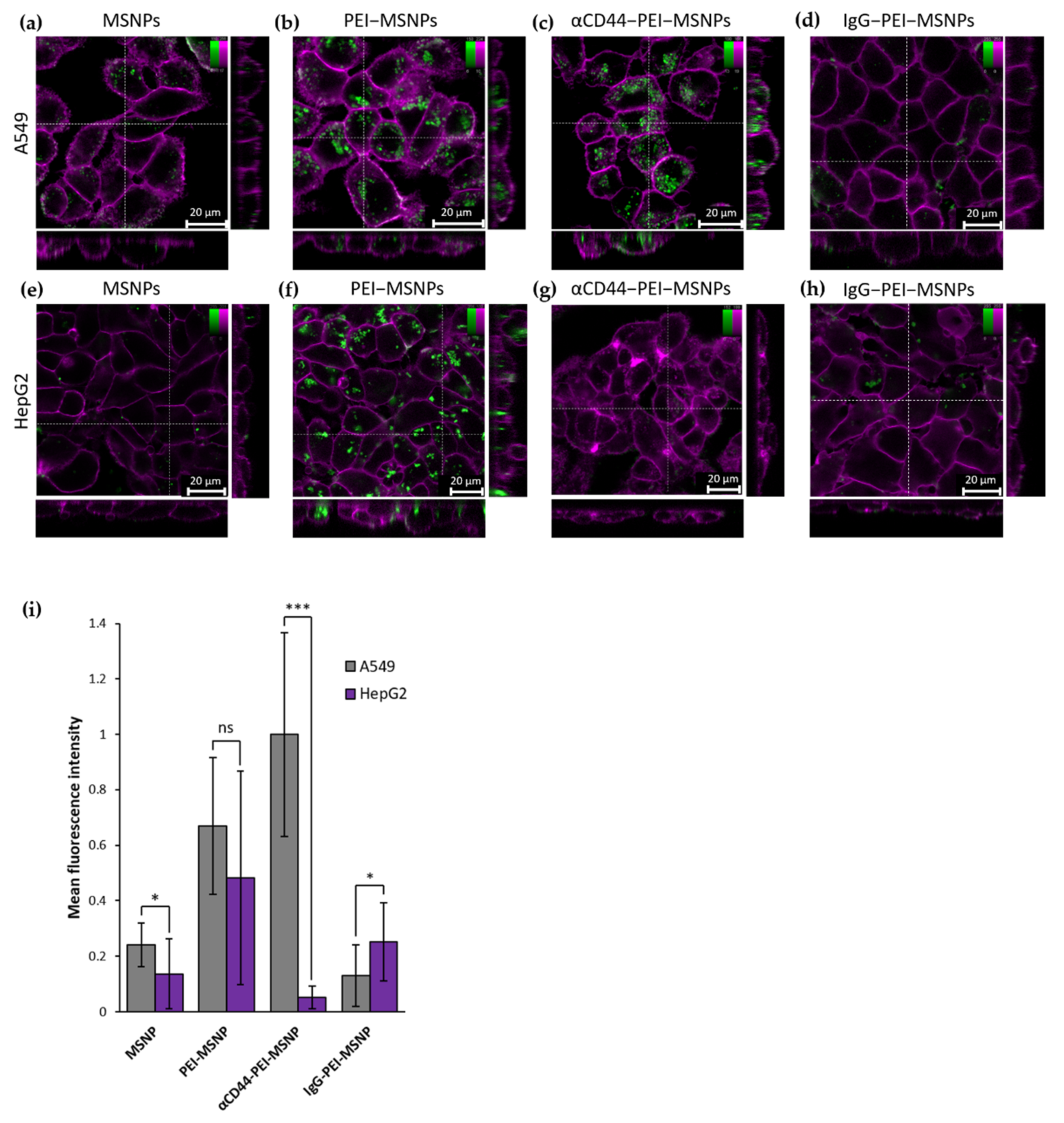
3.2.1. Targeting Capability of αCD44-PEI-MSNPs
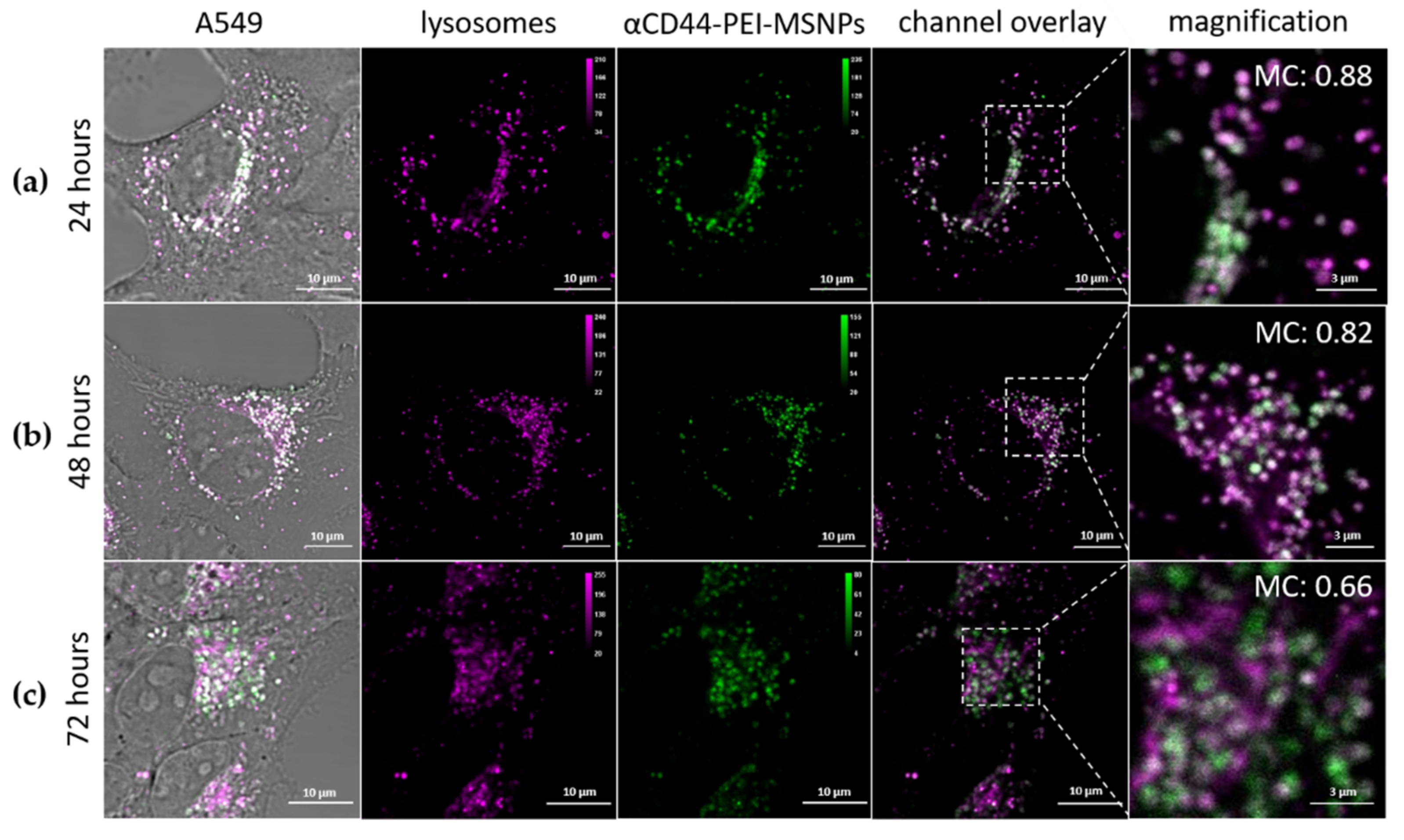
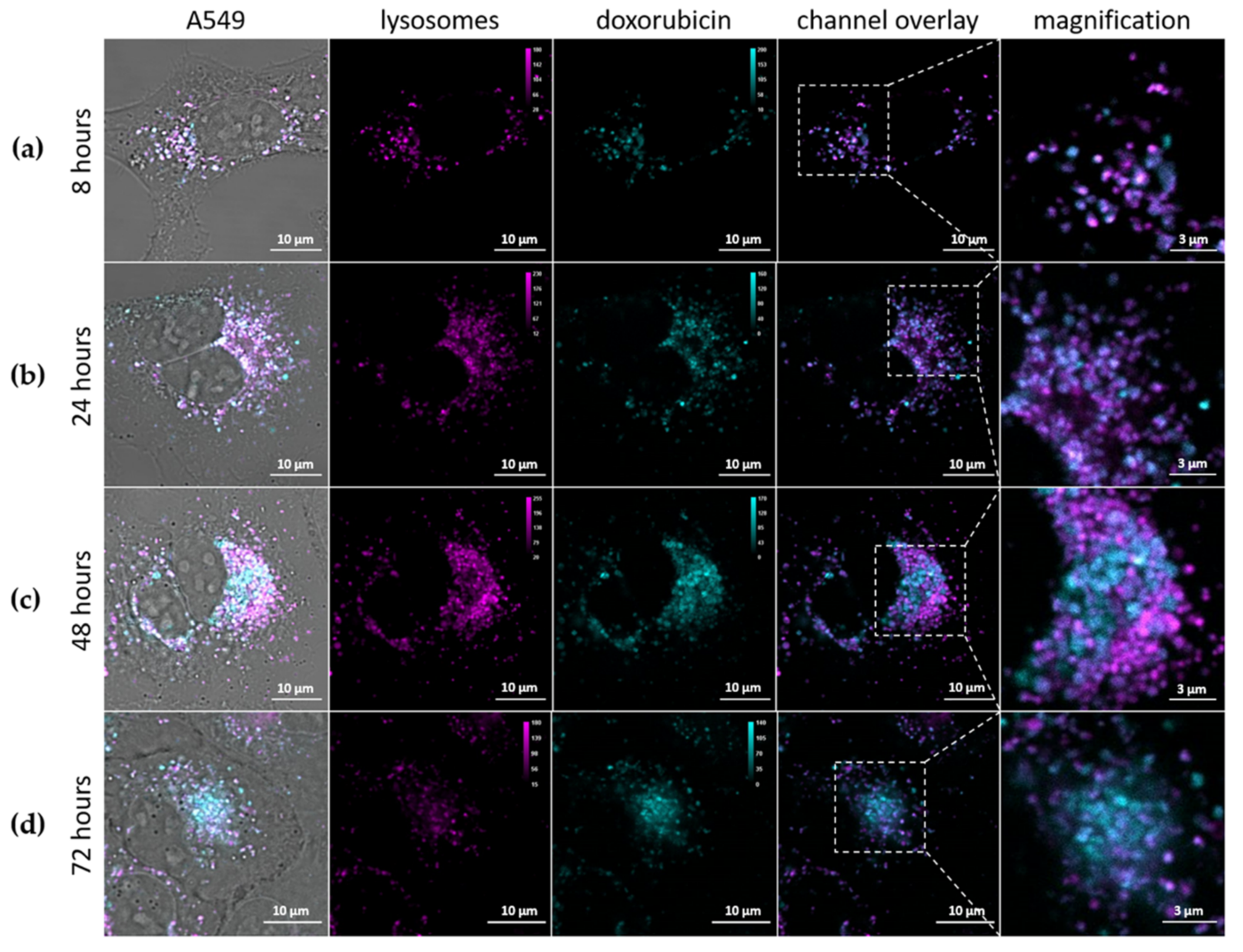
3.2.2. Intracellular Trafficking
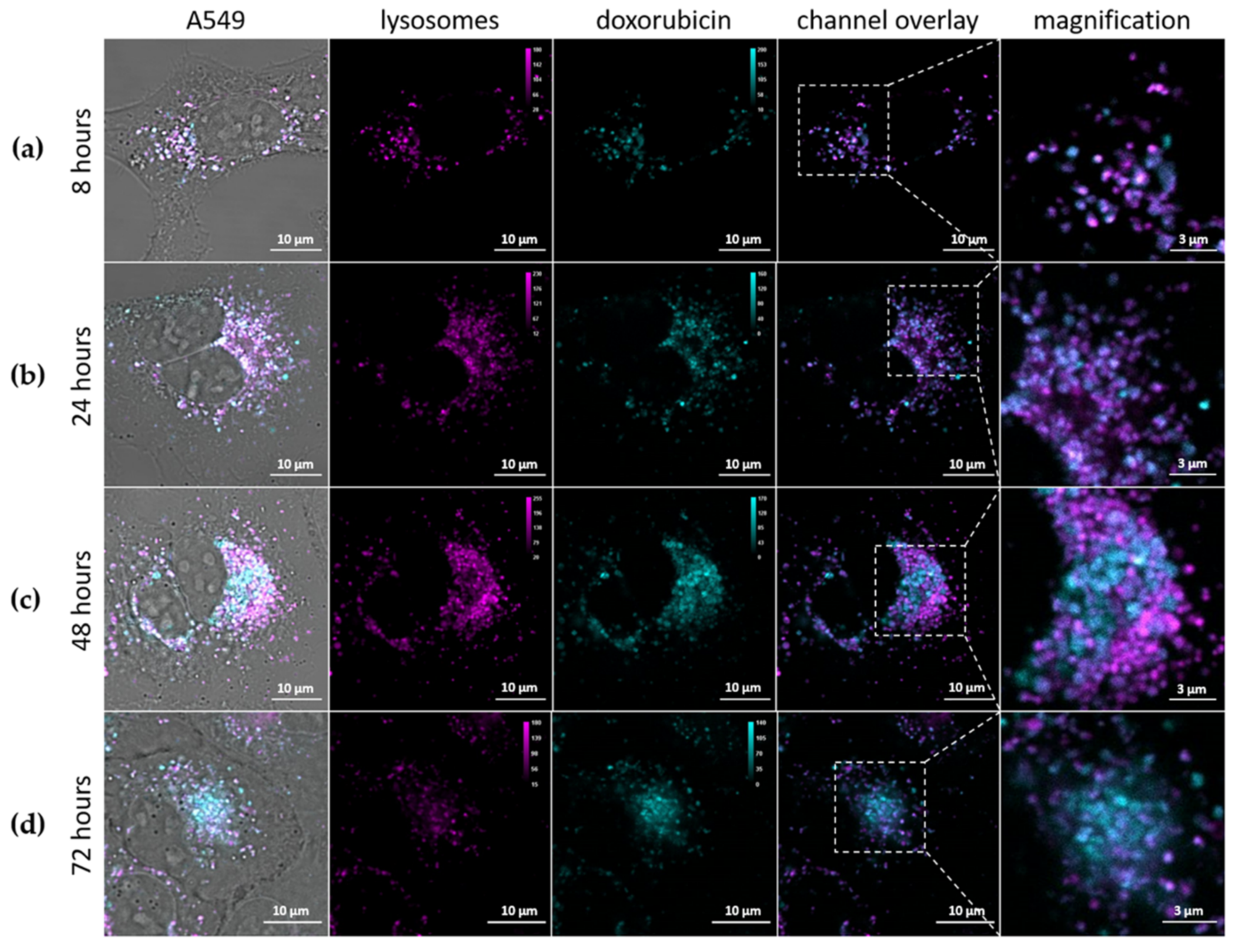
3.2.3. Intracellular Release of Doxorubicin
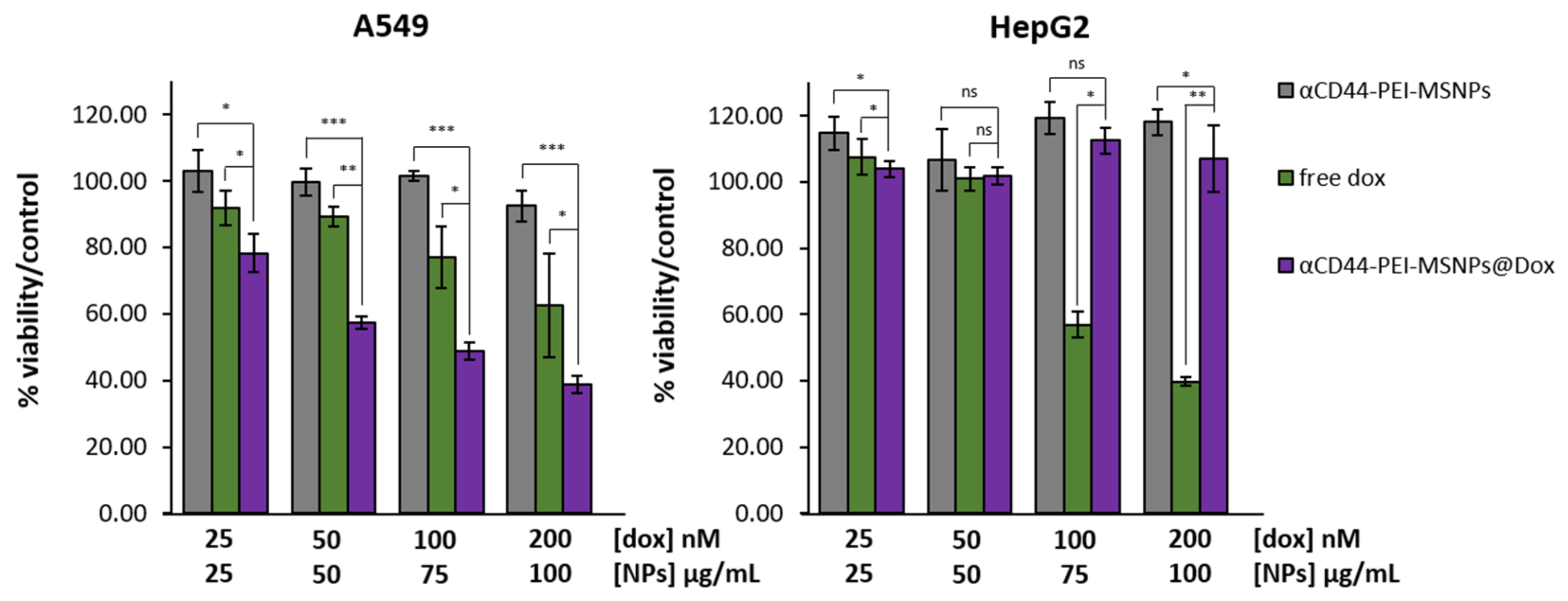
3.2.4. Cytotoxicity Studies
3.3. The αEGFR-Conjugated Nanoparticles
3.3.1. Labelling of the EGFR Antibodies
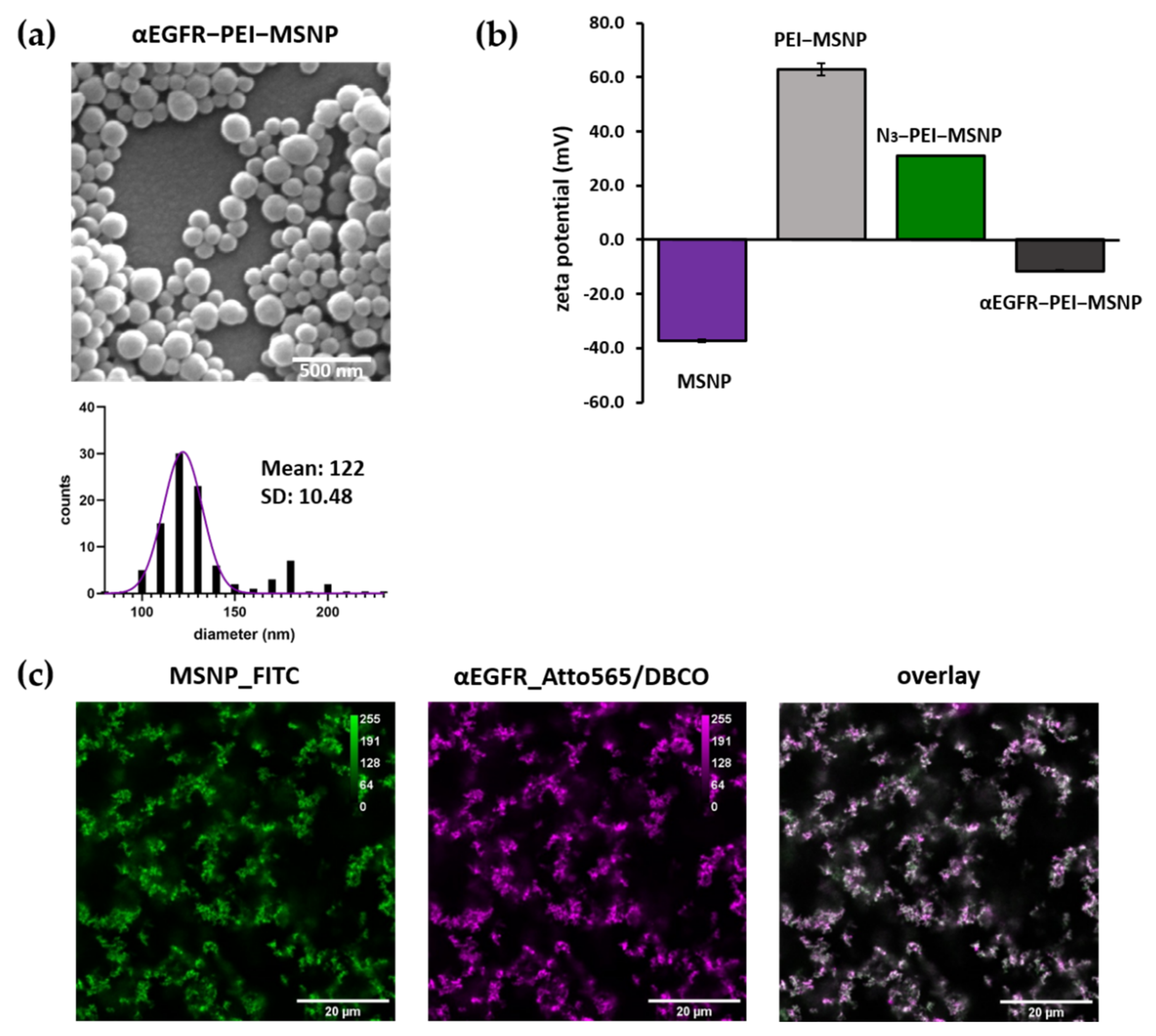
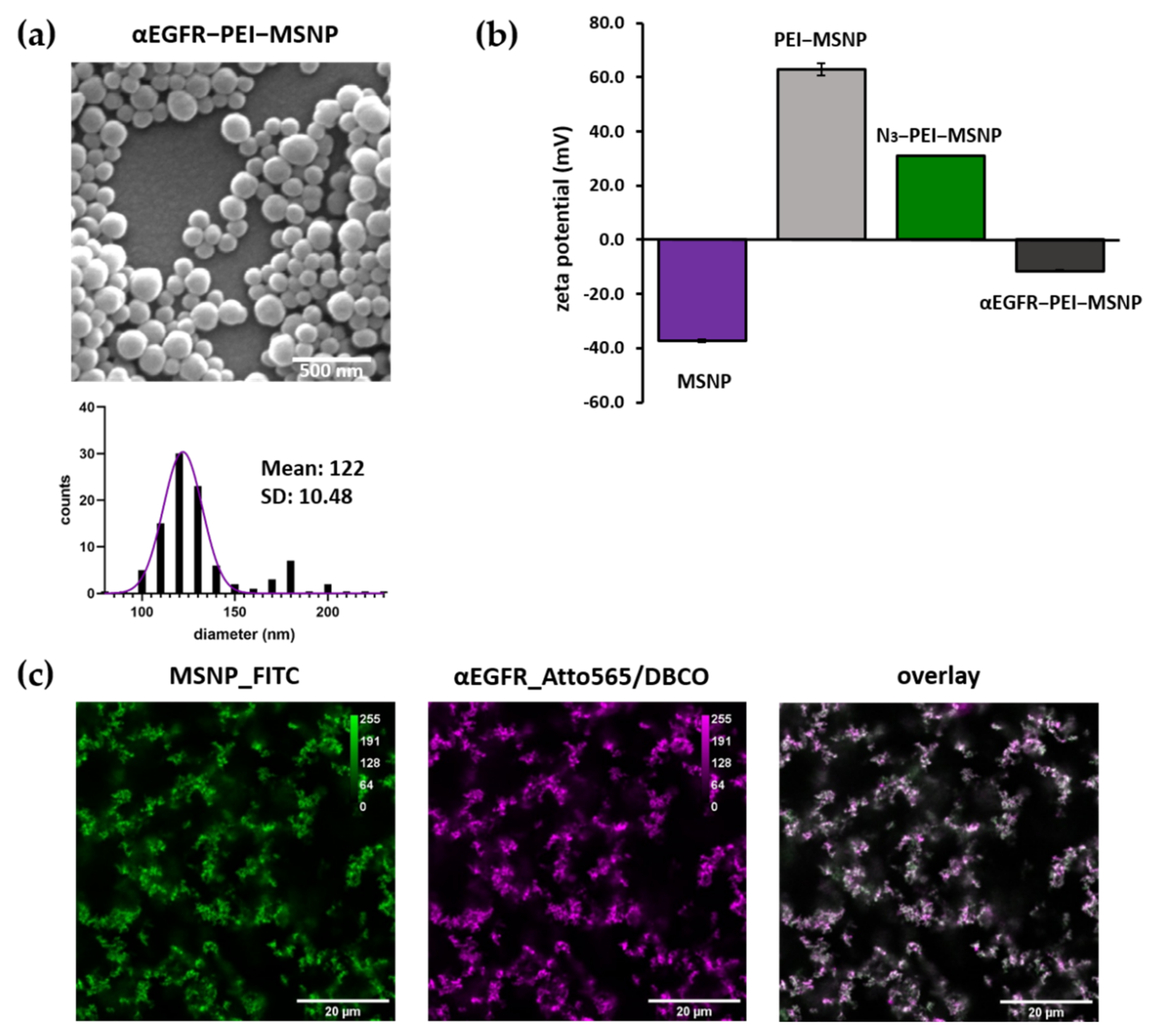
3.3.2. Synthesis and Characterization of αEGFR-Conjugated MSNPs
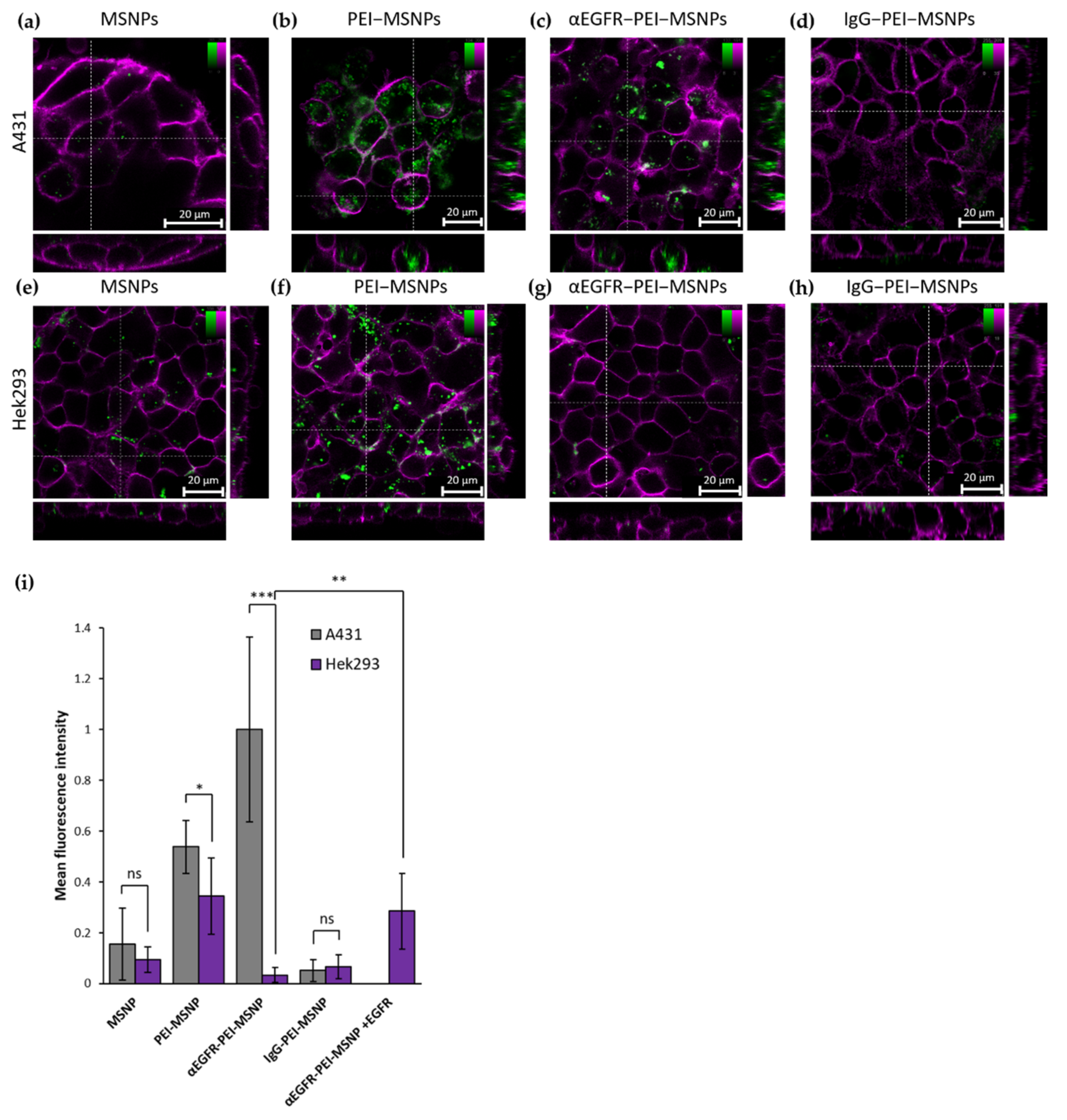
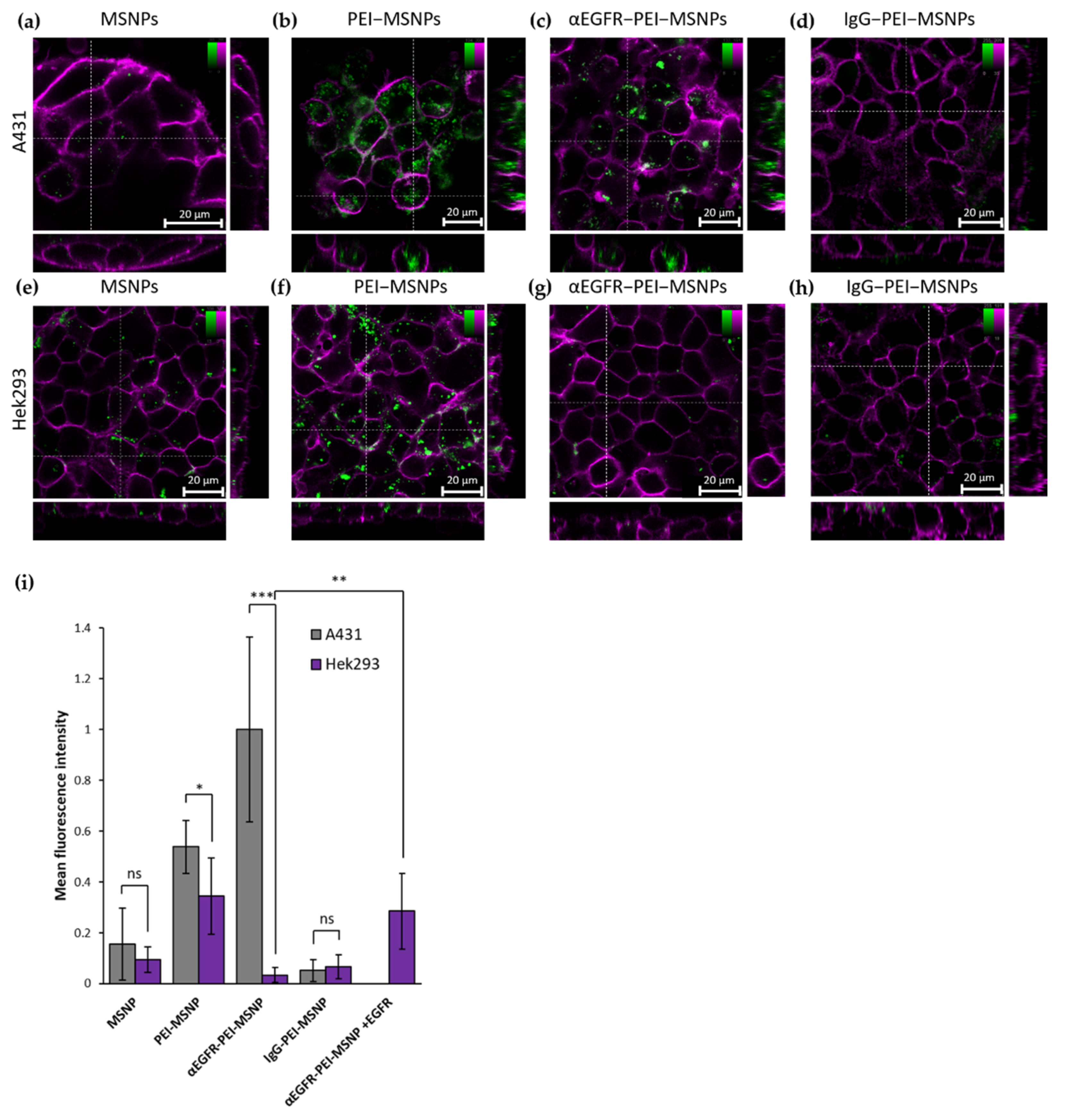
3.3.3. Targeting Capability of αEGFR-PEI-MSNPs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sriraman, S.K.; Aryasomayajula, B.; Torchilin, V.P. Barriers to Drug Delivery in Solid Tumors. Tissue Barriers 2014, 2, e29528. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.-J.; Yoon, I.-S.; Yoon, H.Y.; Koo, H.; Jin, Y.-J.; Ko, S.-H.; Shim, J.S.; Kim, K.; Kwon, I.C.; Kim, D.D. Polyethylene Glycol-Conjugated Hyaluronic Acid-Ceramide Self-Assembled Nanoparticles for Targeted Delivery of Doxorubicin. Biomaterials 2012, 33, 1190–1200. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Shi, P.; Zhao, G.; Xu, J.; Peng, W.; Zhang, J.; Zhang, G.; Wang, X.; Dong, Z.; Chen, F.; et al. Targeting Cancer Stem Cell Pathways for Cancer Therapy. Signal Transduct. Target. Ther. 2020, 5, 1–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greish, K. Enhanced Permeability and Retention (EPR) Effect for Anticancer Nanomedicine Drug Targeting. Methods Mol. Biol. 2010, 624, 25–37. [Google Scholar] [CrossRef]
- Zwicke, G.L.; Mansoori, G.A.; Jeffery, C.J. Utilizing the Folate Receptor for Active Targeting of Cancer Nanotherapeutics. Nano Rev. 2012, 3, 18496. [Google Scholar] [CrossRef]
- Akhter, M.H.; Beg, S.; Tarique, M.; Malik, A.; Afaq, S.; Choudhry, H.; Hosawi, S. Receptor-Based Targeting of Engineered Nanocarrier against Solid Tumors: Recent Progress and Challenges Ahead. Biochim. Biophys. Acta Gen. Subj. 2020, 1865, 129777. [Google Scholar] [CrossRef]
- Wang, A.Z. EPR or no EPR? The Billion-Dollar Question. Sci. Transl. Med. 2015, 7, ec112–ec294. [Google Scholar] [CrossRef]
- Nel, A.; Ruoslahti, E.; Meng, H. New Insights into “Permeability” as in the Enhanced Permeability and Retention Effect of Cancer Nanotherapeutics. ACS Nano 2017, 11, 9567–9569. [Google Scholar] [CrossRef] [PubMed]
- Dai, Q.; Wilhelm, S.; Ding, D.; Syed, A.M.; Sindhwani, S.; Zhang, Y.; Chen, Y.Y.; MacMillan, P.; Chan, W.C. Quantifying the Ligand-Coated Nanoparticle Delivery to Cancer Cells in Solid Tumors. ACS Nano 2018, 12, 8423–8435. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lee, M.-Y.; Hogg, M.G.; Dordick, J.S.; Sharfstein, S.T. Gene Delivery in Three-Dimensional Cell Cultures by Superparamagnetic Nanoparticles. ACS Nano 2010, 4, 4733–4743. [Google Scholar] [CrossRef]
- Wilhelm, S.; Tavares, A.J.; Dai, Q.; Ohta, S.; Audet, J.; Dvorak, H.F.; Chan, W.C. Analysis of Nanoparticle Delivery to Tumours. Nat. Rev. Mater. 2016, 1, 16014. [Google Scholar] [CrossRef]
- Ukidve, A.; Cu, K.; Kumbhojkar, N.; Lahann, J.; Mitragotri, S. Overcoming Biological Barriers to Improve Solid Tumor Immunotherapy. Drug Deliv. Transl. Res. 2021, 11, 2276–2301. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Chen, X.; Cao, J.; Gao, H. Overcoming the Biological Barriers in the Tumor Microenvironment for Improving Drug Delivery and Efficacy. J. Mater. Chem. B 2020, 8, 6765–6781. [Google Scholar] [CrossRef] [PubMed]
- Van Zundert, I.; Fortuni, B.; Rocha, S. From 2D to 3D Cancer Cell Models—The Enigmas of Drug Delivery Research. Nanomaterials 2020, 10, 2236. [Google Scholar] [CrossRef]
- Sherman, M.H.; Yu, R.T.; Engle, D.D.; Ding, N.; Atkins, A.R.; Tiriac, H.; Collisson, E.A.; Connor, F.; Van Dyke, T.; Kozlov, S.; et al. Vitamin D Receptor-Mediated Stromal Reprogramming Suppresses Pancreatitis and Enhances Pancreatic Cancer Therapy. Cell 2014, 159, 80–93. [Google Scholar] [CrossRef] [Green Version]
- Bertrand, N.; Wu, J.; Xu, X.; Kamaly, N.; Farokhzad, O.C. Cancer Nanotechnology: The Impact of Passive and Active Targeting in the Era of Modern Cancer Biology. Adv. Drug Deliv. Rev. 2014, 66, 2–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhirde, A.A.; Patel, V.; Gavard, J.; Zhang, G.; Sousa, A.A.; Masedunskas, A.; Leapman, R.D.; Weigert, R.; Gutkind, J.S.; Rusling, J.F. Targeted Killing of Cancer Cells in vivo and in vitro with EGF-Directed Carbon Nanotube-based Drug Delivery. ACS Nano 2009, 3, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Gan, C.W.; Feng, S.-S. Transferrin-Conjugated Nanoparticles of Poly(Lactide)-D-Alpha-Tocopheryl Polyethylene Glycol Succinate Diblock Copolymer for Targeted Drug Delivery across the Blood-Brain Barrier. Biomaterials 2010, 31, 7748–7757. [Google Scholar] [CrossRef]
- Gerber, D.E. Targeted Therapies: A New Generation of Cancer Treatments. Am. Fam. Physician 2008, 77, 311–319. [Google Scholar]
- Gaspar, V.M.; Costa, E.C.; Queiroz, J.A.; Pichon, C.; Sousa, F.; Correia, I.J. Folate-Targeted Multifunctional Amino Acid-Chitosan Nanoparticles for Improved Cancer Therapy. Pharm. Res. 2015, 32, 562–577. [Google Scholar] [CrossRef]
- Wang, X.; Li, J.; Wang, Y.; Koenig, L.; Gjyrezi, A.; Giannakakou, P.; Shin, E.H.; Tighiouart, M.; Chen, Z.; Nie, S.; et al. A Folate Receptor-Targeting Nanoparticle Minimizes Drug Resistance in a Human Cancer Model. ACS Nano 2011, 5, 6184–6194. [Google Scholar] [CrossRef]
- Hsu, J.L.; Hung, M.-C. The Role of HER2, EGFR, and Other Receptor Tyrosine Kinases in Breast Cancer. Cancer Metastasis Rev. 2016, 35, 575–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lumachi, F.; Brunello, A.; Maruzzo, M.; Basso, U.; Basso, S.M.M. Treatment of Estrogen Receptor-Positive Breast Cancer. Curr. Med. Chem. 2013, 20, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Perou, C.M.; Sørlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular Portraits of Human Breast Tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef]
- Senter, P.D.; Sievers, E.L. The Discovery and Development of Brentuximab Vedotin for Use in Relapsed Hodgkin Lymphoma and Systemic Anaplastic Large Cell Lymphoma. Nat. Biotechnol. 2012, 30, 631–637. [Google Scholar] [CrossRef] [PubMed]
- Amiri-Kordestani, L.; Blumenthal, G.M.; Xu, Q.C.; Zhang, L.; Tang, S.W.; Ha, L.; Weinberg, W.C.; Chi, B.; Candau-Chacon, R.; Hughes, P.; et al. FDA Approval: Ado-Trastuzumab Emtansine for the Treatment of Patients with HER2-Positive Metastatic Breast Cancer. Clin. Cancer Res. 2014, 20, 4436–4441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wynne, J.; Wright, D.; Stock, W. Inotuzumab: From Preclinical Development to Success in B-cell Acute Lymphoblastic Leukemia. Blood Adv. 2019, 3, 96–104. [Google Scholar] [CrossRef]
- Hafeez, U.; Parakh, S.; Gan, H.K.; Scott, A.M. Antibody–Drug Conjugates for Cancer Therapy. Molecules 2020, 25, 4764. [Google Scholar] [CrossRef] [PubMed]
- Hammood, M.; Craig, A.W.; Leyton, J.V. Impact of Endocytosis Mechanisms for the Receptors Targeted by the Currently Approved Antibody-Drug Conjugates (ADCs)—A Necessity for Future ADC Research and Development. Pharmaceuticals 2021, 14, 674. [Google Scholar] [CrossRef] [PubMed]
- Johnston, M.C.; Scott, C.J. Antibody Conjugated Nanoparticles as a Novel Form of Antibody Drug Conjugate Chemotherapy. Drug Discov. Today Technol. 2018, 30, 63–69. [Google Scholar] [CrossRef]
- Juan, A.; Cimas, F.J.; Bravo, I.; Pandiella, A.; Ocaña, A.; Alonso-Moreno, C. An Overview of Antibody Conjugated Polymeric Nanoparticles for Breast Cancer Therapy. Pharmaceutics 2020, 12, 802. [Google Scholar] [CrossRef] [PubMed]
- Yu, K.; Zhao, J.; Zhang, Z.; Gao, Y.; Zhou, Y.; Teng, L.; Li, Y. Enhanced Delivery of Paclitaxel Using Electrostatically-Conjugated Herceptin-Bearing PEI/PLGA Nanoparticles against HER-Positive Breast Cancer Cells. Int. J. Pharm. 2016, 497, 78–87. [Google Scholar] [CrossRef]
- Mi, Y.; Huang, Y.; Deng, J. The Enhanced Delivery of Salinomycin to CD133+ Ovarian Cancer Stem Cells through CD133 Antibody Conjugation with Poly(Lactic-Co-Glycolic Acid)-Poly(Ethylene Glycol) Nanoparticles. Oncol. Lett. 2018, 15, 6611–6621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varshosaz, J.; Davoudi, M.A.; Rasoul-Amini, S. Docetaxel-Loaded Nanostructured Lipid Carriers Functionalized with Trastuzumab (Herceptin) for HER2-Positive Breast Cancer Cells. J. Liposome Res. 2018, 28, 285–295. [Google Scholar] [CrossRef]
- Chen, S.; Florinas, S.; Teitgen, A.; Xu, Z.-Q.; Gao, C.; Wu, H.; Kataoka, K.; Cabral, H.; Christie, R.J. Controlled Fab Installation onto Polymeric Micelle Nanoparticles for Tuned Bioactivity. Sci. Technol. Adv. Mater. 2017, 18, 666–680. [Google Scholar] [CrossRef]
- Wartlick, H.; Michaelis, K.; Balthasar, S.; Strebhardt, K.; Kreuter, J.; Langer, K. Highly Specific HER2-Mediated Cellular Uptake of Antibody-Modified Nanoparticles in Tumour Cells. J. Drug Target. 2004, 12, 461–471. [Google Scholar] [CrossRef]
- Goossens, J.; Sein, H.; Lu, S.; Radwanska, M.; Muyldermans, S.; Sterckx, Y.G.-J.; Magez, S. Functionalization of Gold Nanoparticles with Nanobodies through Physical Adsorption. Anal. Methods 2017, 9, 3430–3440. [Google Scholar] [CrossRef]
- Choi, J.-S.; Jang, W.S.; Park, J.-S. Comparison of Adsorption and Conjugation of Herceptin on Poly(Lactic-Co-Glycolic Acid) Nanoparticles-Effect on Cell Internalization in Breast Cancer Cells. Mater. Sci. Eng. C Mater. Biol. Appl. 2018, 92, 496–507. [Google Scholar] [CrossRef]
- Jain, A.; Cheng, K. The Principles and Applications of Avidin-Based Nanoparticles in Drug Delivery and Diagnosis. J. Control. Release 2017, 245, 27–40. [Google Scholar] [CrossRef] [Green Version]
- Sperling, R.A.; Parak, W.J. Surface Modification, Functionalization and Bioconjugation of Colloidal Inorganic Nanoparticles. Philos. Trans. A Math. Phys. Eng. Sci. 2010, 368, 1333–1383. [Google Scholar] [CrossRef]
- Kim, C.; Galloway, J.F.; Lee, K.H.; Searson, P.C. Universal Antibody Conjugation to Nanoparticles Using the Fcγ Receptor I (FcγRI): Quantitative Profiling Of Membrane Biomarkers. Bioconjug Chem. 2014, 25, 1893–1901. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Koo, H. Biomedical Applications of Copper-Free Click Chemistry: In vitro, in vivo, and ex vivo. Chem. Sci. 2019, 10, 7835–7851. [Google Scholar] [CrossRef] [Green Version]
- Egorova, K.S.; Ananikov, V.P. Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar] [CrossRef] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging Functions of the EGFR in Cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef] [PubMed]
- Naor, D.; Sionov, R.V.; Ish-Shalom, D. CD44: Structure, Function, and Association with the Malignant Process. Adv. Cancer Res. 1997, 71, 241–319. [Google Scholar] [CrossRef] [PubMed]
- Slowing, I.I.; Trewyn, B.G.; Giri, S.; Lin, V.S.-Y. Mesoporous Silica Nanoparticles for Drug Delivery and Biosensing Applications. Adv. Funct. Mater. 2007, 17, 1225–1236. [Google Scholar] [CrossRef]
- Wang, S. Ordered Mesoporous Materials for Drug Delivery. Microporous Mesoporous Mater. 2009, 117, 1–9. [Google Scholar] [CrossRef]
- Garrido-Cano, I.; Candela-Noguera, V.; Herrera, G.; Cejalvo, J.M.; Lluch, A.; Marcos, M.D.; Sancenon, F.; Eroles, P.; Martínez-Máñez, R. Biocompatibility and Internalization Assessment of Bare and Functionalised Mesoporous Silica Nanoparticles. Microporous Mesoporous Mater. 2021, 310, 110593. [Google Scholar] [CrossRef]
- Fortuni, B.; Inose, T.; Ricci, M.; Fujita, Y.; Van Zundert, I.; Masuhara, A.; Fron, E.; Mizuno, H.; Latterini, L.; Rocha, S.; et al. Polymeric Engineering of Nanoparticles for Highly Efficient Multifunctional Drug Delivery Systems. Sci. Rep. 2019, 9, 2666. [Google Scholar] [CrossRef]
- Thirukkumaran, O.M.; Kluba, M.; Hofkens, J.; Mizuno, H. Autophosphorylation of EGFR at Y954 Facilitated Homodimerization and Enhanced Downstream Signals. Biophys. J. 2020, 119, 2127–2137. [Google Scholar] [CrossRef]
- Taemaitree, F.; Fortuni, B.; Koseki, Y.; Fron, E.; Rocha, S.; Hofkens, J.; Uji-i, H.; Inose, T.; Kasai, H. FRET-Based Intracellular Investigation of Nanoprodrugs toward Highly Efficient Anticancer Drug Delivery. Nanoscale 2020, 12, 16710–16715. [Google Scholar] [CrossRef]
- Hammink, R.; Weiden, J.; Voerman, D.; Popelier, C.; Eggermont, L.J.; Schluck, M.; Figdor, C.G.; Verdoes, M. Semiflexible Immunobrushes Induce Enhanced T Cell Activation and Expansion. ACS Appl. Mater. Interfaces 2021, 13, 16007–16018. [Google Scholar] [CrossRef] [PubMed]
- Wiener, J.; Kokotek, D.; Rosowski, S.; Lickert, H.; Meier, M. Preparation of Single- and Double-Oligonucleotide Antibody Conjugates and Their Application for Protein Analytics. Sci. Rep. 2020, 10, 1457. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.; Yang, J.; Li, X.; Zhou, L.; Zhang, R.; Li, W.; Chen, L.; Wang, R.; Zhang, F.; Zhao, D. Biphase Stratification Approach to Three-Dimensional Dendritic Biodegradable Mesoporous Silica Nanospheres. Nano Lett. 2014, 14, 923–932. [Google Scholar] [CrossRef] [PubMed]
- Nečas, D.; Klapetek, P. Gwyddion: An Open-Source Software for SPM Data Analysis. Open Phys. 2012, 10, 181–188. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An Open-Source Platform for Biological-Image Analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolte, S.; Cordelières, F.P. A Guided Tour into Subcellular Colocalization Analysis in Light Microscopy. J. Microsc. 2006, 224, 213–232. [Google Scholar] [CrossRef]
- Manders, E.M.M.; Verbeek, F.J.; Aten, J.A. Measurement of Co-Localization of Objects in Dual-Colour Confocal Images. J. Microsc. 1993, 169, 375–382. [Google Scholar] [CrossRef]
- Goedhart, J. Plots of Differences—A Web App for the Quantitative Comparison of Unpaired Data. BioRxiv 2019, 578575. [Google Scholar] [CrossRef] [Green Version]
- Penno, M.B.; August, J.T.; Baylin, S.B.; Mabry, M.; Linnoila, R.I.; Lee, V.S.; Croteau, D.; Yang, X.L.; Rosada, C. Expression of CD44 in Human Lung Tumors. Cancer Res. 1994, 54, 1381–1387. [Google Scholar]
- Dalerba, P.; Dylla, S.J.; Park, I.-K.; Liu, R.; Wang, X.; Cho, R.W.; Hoey, T.; Gurney, A.; Huang, E.H.; Simeone, D.M.; et al. Phenotypic Characterization of Human Colorectal Cancer Stem Cells. Proc. Natl. Acad. Sci. USA 2007, 104, 10158–10163. [Google Scholar] [CrossRef] [Green Version]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective Identification of Tumorigenic Breast Cancer Cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef] [Green Version]
- Arabi, L.; Badiee, A.; Mosaffa, F.; Jaafari, M.R. Targeting CD44 Expressing Cancer Cells with Anti-CD44 Monoclonal Antibody Improves Cellular Uptake and Antitumor Efficacy of Liposomal Doxorubicin. J. Control. Release 2015, 220, 275–286. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Stower, M.J.; Maitland, N.J. Prospective Identification of Tumorigenic Prostate Cancer Stem Cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef] [Green Version]
- Ayob, A.Z.; Ramasamy, T.S. Cancer Stem Cells as Key Drivers of Tumour Progression. J. Biomed. Sci. 2018, 25, 20. [Google Scholar] [CrossRef] [PubMed]
- Arpicco, S.; Lerda, C.; Dalla Pozza, E.; Costanzo, C.; Tsapis, N.; Stella, B.; Donadelli, M.; Dando, I.; Fattal, E.; Cattel, L.; et al. Hyaluronic Acid-Coated Liposomes for Active Targeting of Gemcitabine. Eur. J. Pharm. Biopharm. 2013, 85, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, Z.; Lin, Y.; Yin, M.; Ren, J.; Qu, X. Bioresponsive Hyaluronic Acid-Capped Mesoporous Silica Nanoparticles for Targeted Drug Delivery. Chemistry 2013, 19, 1778–1783. [Google Scholar] [CrossRef] [PubMed]
- Wei, J.; Sun, J.; Liu, Y. Enhanced Targeting of Prostate Cancer-Initiating Cells by Salinomycin-Encapsulated Lipid-PLGA Nanoparticles Linked with CD44 Antibodies. Oncol. Lett. 2019, 17, 4024–4033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Su, W.; Liu, Z.; Zhou, M.; Chen, S.; Chen, Y.; Lu, D.; Liu, Y.; Fan, Y.; Zheng, Y.; et al. CD44 Antibody-Targeted Liposomal Nanoparticles for Molecular Imaging and Therapy of Hepatocellular Carcinoma. Biomaterials 2012, 33, 5107–5114. [Google Scholar] [CrossRef]
- Su, Z.; Liu, D.; Chen, L.; Zhang, J.; Ru, L.; Chen, Z.; Gao, Z.; Wang, X. CD44-Targeted Magnetic Nanoparticles Kill Head And Neck Squamous Cell Carcinoma Stem Cells In An Alternating Magnetic Field. Int. J. Nanomed. 2019, 14, 7549–7560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, Å.; Ljunglöf, A.; Lindblom, H. One-Step Purification of Monoclonal IgG Antibodies from Mouse Ascites: An Evaluation of Different Adsorption Techniques Using High Performance Liquid Chromatography. J. Immunol. Methods 1988, 115, 79–88. [Google Scholar] [CrossRef]
- Zhang, L.; Hu, D.; Salmain, M.; Liedberg, B.; Boujday, S. Direct Quantification of Surface Coverage of Antibody in IgG-Gold Nanoparticles Conjugates. Talanta 2019, 204, 875–881. [Google Scholar] [CrossRef] [PubMed]
- Gregory, J. Monitoring Particle Aggregation Processes. Adv. Colloid Interface Sci. 2009, 147–148, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Foster, K.A.; Oster, C.G.; Mayer, M.M.; Avery, M.L.; Audus, K.L. Characterization of the A549 Cell Line as a Type II Pulmonary Epithelial Cell Model for Drug Metabolism. Exp. Cell. Res. 1998, 243, 359–366. [Google Scholar] [CrossRef] [PubMed]
- Aden, D.P.; Fogel, A.; Plotkin, S.; Damjanov, I.; Knowles, B.B. Controlled Synthesis of HBsAg in a Differentiated Human Liver Carcinoma-Derived Cell Line. Nature 1979, 282, 615–616. [Google Scholar] [CrossRef]
- Arvizo, R.R.; Miranda, O.R.; Thompson, M.A.; Pabelick, C.M.; Bhattacharya, R.; Robertson, J.D.; Rotello, V.M.; Prakash, Y.S.; Mukherjee, P. Effect of Nanoparticle Surface Charge at the Plasma Membrane and Beyond. Nano Lett. 2010, 10, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, T.; Kovochich, M.; Liong, M.; Meng, H.; Kabehie, S.; George, S.; Zink, J.I.; Nel, A.E. Polyethyleneimine Coating Enhances the Cellular Uptake of Mesoporous Silica Nanoparticles and Allows Safe Delivery of siRNA and DNA Constructs. ACS Nano 2009, 3, 3273–3286. [Google Scholar] [CrossRef]
- Shao, J.; Liang, R.; Ding, D.; Zheng, X.; Zhu, X.; Hu, S.; Wei, H.; Wei, B. A Smart Multifunctional Nanoparticle for Enhanced Near-Infrared Image-Guided Photothermal Therapy Against Gastric Cancer. IJN 2021, 16, 2897–2915. [Google Scholar] [CrossRef]
- Somasunderam, A.; Thiviyanathan, V.; Tanaka, T.; Li, X.; Neerathilingam, M.; Lokesh, G.L.R.; Mann, A.; Peng, Y.; Ferrari, M.; Klostergaard, J.; et al. Combinatorial Selection of DNA Thioaptamers Targeted to the HA Binding Domain of Human CD44. Biochemistry 2010, 49, 9106–9112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouellette, M.M.; McDaniel, L.D.; Wright, W.E.; Shay, J.W.; Schultz, R.A. The Establishment of Telomerase-Immortalized Cell Lines Representing Human Chromosome Instability Syndromes. Hum. Mol. Genet. 2000, 9, 403–411. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Yasukawa, M.; Nguyen, C.; Kasim, V.; Maida, Y.; Possemato, R.; Shibata, T.; Ligon, K.L.; Fukami, K.; Hahn, W.C.; et al. Maintenance of Tumor Initiating Cells of Defined Genetic Composition by Nucleostemin. Proc. Natl. Acad. Sci. USA 2011, 108, 20388–20393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andrian, T.; Riera, R.; Pujals, S.; Albertazzi, L. Nanoscopy for Endosomal Escape Quantification. Nanoscale Adv. 2021, 3, 10–23. [Google Scholar] [CrossRef]
- Selby, L.I.; Cortez-Jugo, C.M.; Such, G.K.; Johnston, A.P.R. Nanoescapology: Progress toward Understanding the Endosomal Escape of Polymeric Nanoparticles. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2017, 9, e1452. [Google Scholar] [CrossRef]
- Degors, I.M.S.; Wang, C.; Rehman, Z.U.; Zuhorn, I.S. Carriers Break Barriers in Drug Delivery: Endocytosis and Endosomal Escape of Gene Delivery Vectors. Acc. Chem. Res. 2019, 52, 1750–1760. [Google Scholar] [CrossRef] [Green Version]
- Tacar, O.; Sriamornsak, P.; Dass, C.R. Doxorubicin: An Update on Anticancer Molecular Action, Toxicity and Novel Drug Delivery Systems. J. Pharm. Pharmacol. 2013, 65, 157–170. [Google Scholar] [CrossRef]
- Kralj, S.; Rojnik, M.; Kos, J.; Makovec, D. Targeting EGFR-Overexpressed A431 Cells with EGF-Labeled Silica-Coated Magnetic Nanoparticles. J. Nanopart. Res. 2013, 15, 1666. [Google Scholar] [CrossRef]
- Patel, J.; Amrutiya, J.; Bhatt, P.; Javia, A.; Jain, M.; Misra, A. Targeted Delivery of Monoclonal Antibody Conjugated Docetaxel Loaded PLGA Nanoparticles into EGFR Overexpressed Lung Tumour Cells. J. Microencapsul. 2018, 35, 204–217. [Google Scholar] [CrossRef]
- Kuroda, S.; Tam, J.; Roth, J.A.; Sokolov, K.; Ramesh, R. EGFR-Targeted Plasmonic Magnetic Nanoparticles Suppress Lung Tumor Growth by Abrogating G2/M Cell-Cycle Arrest and Inducing DNA Damage. IJN 2014, 9, 3825–3839. [Google Scholar] [CrossRef] [Green Version]
- Acharya, S.; Dilnawaz, F.; Sahoo, S.K. Targeted Epidermal Growth Factor Receptor Nanoparticle Bioconjugates for Breast Cancer Therapy. Biomaterials 2009, 30, 5737–5750. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Xu, X.; Luo, W.-J. Construction of a High-EGFR Expression Cell Line and Its Biological Properties Comparing with A431 Cell. Afr. J. Biotechnol. 2010, 9, 4674–4680. [Google Scholar] [CrossRef]
- Zhang, F.; Wang, S.; Yin, L.; Yang, Y.; Guan, Y.; Wang, W.; Xu, H.; Tao, N. Quantification of Epidermal Growth Factor Receptor Expression Level and Binding Kinetics on Cell Surfaces by Surface Plasmon Resonance Imaging. Anal. Chem. 2015, 87, 9960–9965. [Google Scholar] [CrossRef] [PubMed] [Green Version]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Van Zundert, I.; Bravo, M.; Deschaume, O.; Cybulski, P.; Bartic, C.; Hofkens, J.; Uji-i, H.; Fortuni, B.; Rocha, S. Versatile and Robust Method for Antibody Conjugation to Nanoparticles with High Targeting Efficiency. Pharmaceutics 2021, 13, 2153. https://doi.org/10.3390/pharmaceutics13122153
Van Zundert I, Bravo M, Deschaume O, Cybulski P, Bartic C, Hofkens J, Uji-i H, Fortuni B, Rocha S. Versatile and Robust Method for Antibody Conjugation to Nanoparticles with High Targeting Efficiency. Pharmaceutics. 2021; 13(12):2153. https://doi.org/10.3390/pharmaceutics13122153
Chicago/Turabian StyleVan Zundert, Indra, Maria Bravo, Olivier Deschaume, Pierre Cybulski, Carmen Bartic, Johan Hofkens, Hiroshi Uji-i, Beatrice Fortuni, and Susana Rocha. 2021. "Versatile and Robust Method for Antibody Conjugation to Nanoparticles with High Targeting Efficiency" Pharmaceutics 13, no. 12: 2153. https://doi.org/10.3390/pharmaceutics13122153
APA StyleVan Zundert, I., Bravo, M., Deschaume, O., Cybulski, P., Bartic, C., Hofkens, J., Uji-i, H., Fortuni, B., & Rocha, S. (2021). Versatile and Robust Method for Antibody Conjugation to Nanoparticles with High Targeting Efficiency. Pharmaceutics, 13(12), 2153. https://doi.org/10.3390/pharmaceutics13122153